BIOCHEMISTRY TOPICS
Functional groups
Important organic functional groups for biochemistry
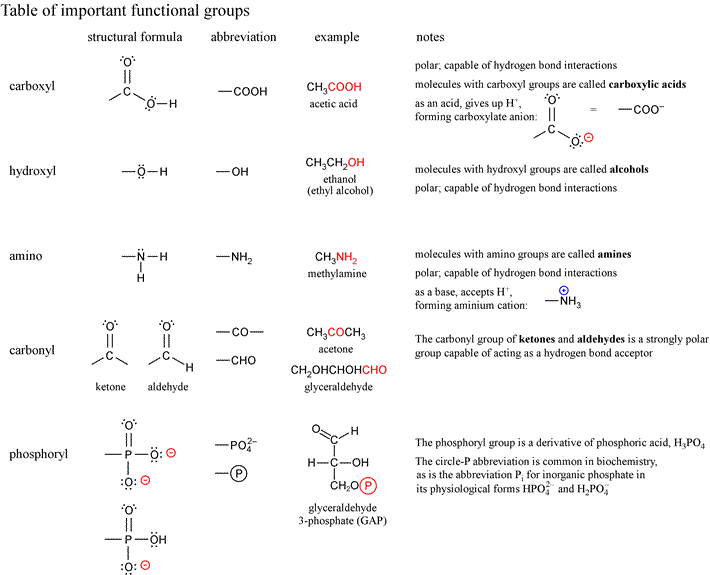
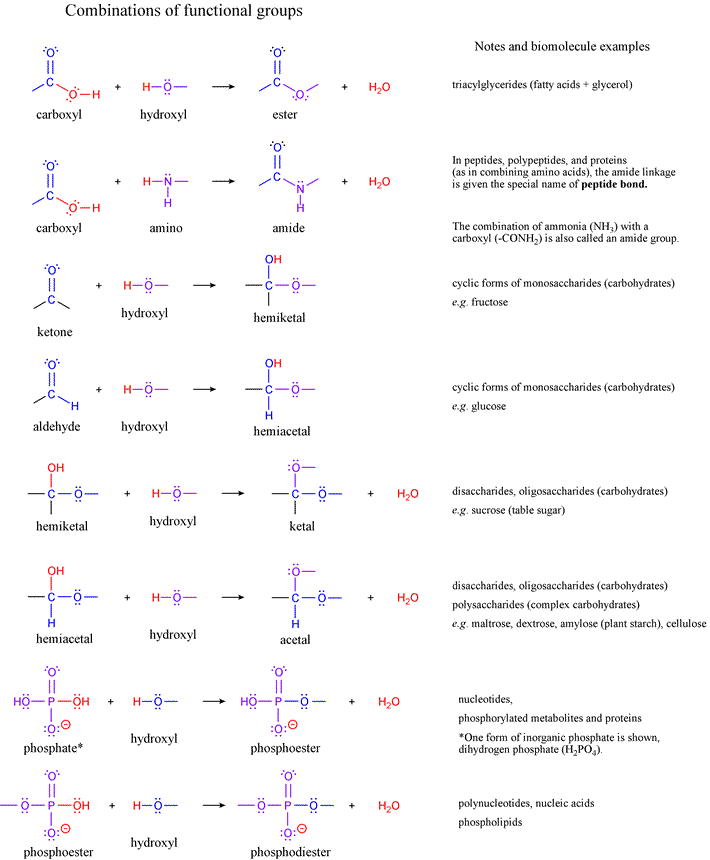
List of functional groups of biomolecules from Lehninger7e, Fig. 1-17
- methyl
- ethyl
- phenyl
- carbonyl (aldehyde)
- carbonyl (ketone)
- carboxyl
- hydroxyl (alcohol)
- enol
- ether
- ester
- acetyl
- anhydride
- amino
- imine
- N-substituted imine (Schiff base)
- guanidinium
- imidazole
- sulfhydryl
- disulfide
- thioester
- phosphoryl
- phosphoanhydride
- mixed anhydride (carboxylic acid and phosphoric acid, a.k.a. acyl phosphate)
Structures. Chymotrypsin. pdb entry 5cha Molecule of the Month. Lysozyme. Molecule of the Month (September 2000). @Protein Data Bank
Related topics pages: bioenergetics |
Related topics pages:
Enzymes. Transferases (Transfer of functional groups) Example : aspartate transcarbamoylase [EC 2.1.3.2]
Equations. The free energy of a difference in electrochemical potential across a membrane is given by
ΔG = RT·ln Q + ZFΔV,
where Q is the ratio of activities of the chemical species on either side of the membrane
- we can take this as dependent on H+ concentrations,
Z is the charge of the species (+1 for H+),
F is the Faraday constant (96.485 kJ·mol−1·V−1),
and ΔV is the electric potential difference, in volts, across the membrane.
For H+, the side of the membrane with the higher concentration (lower pH) will be the more positive
in electric potential. Thus, the two terms in the equation above (considering just H+), will
have the same sign and add together to determine ΔG.
Recall that nuclear magnetic resonance (NMR) is based upon the quantum nature of atomic nuclei;
in particular, a property called "nuclear spin". In the absence of an applied magnetic field,
the two possible spin states of a proton (1H nucleus) are degenerate, i.e. of equal energy.
However, an external magnetic field creates an energy gap ΔE that is proportional to the strength
Ho of the applied field:
ΔE = hνo, where νo = γHo/2π .
In the above relation, γ is the magnetogyric ratio, a constant characteristic of the nucleus being observed in the magnetic field. The equation expresses the condition for a resonant frequency (of electromagnetic radiation) νo, which lies in the radio frequency part of the spectrum. When a photon (quantum of electromagnetic radiation) with the resonant frequency is absorbed by the nucleus in the lower energy state (with the nuclear spin aligned with the direction of the magnetic field, analogous to the alignment of a compass needle to the earth's magnetic north pole), the nuclear spin can be flipped into the higher energy spin state. An NMR spectrometer is capable of measuring this frequency-dependent absorption, and producing a spectrum capturing the signals arising from all the 1H nuclei of an organic molecule, and in principle, from many other isotopic types of nuclei making it possible to study virtually any molecule of interest using NMR.
In "1-D" 1H-NMR for small organic molecules, the different hydrogens are found in differing chemical (electronic) environments, the result being that the magnetic field strength at a particular hydrogen nucleus v aries from the applied field strength by the effects of electrodynamic shielding or deshielding. This gives rise to what is termed chemical shift, δ, which is defined using a reference frequency.
The 1H-NMR spectra for small organic molecules reveal other features besides the chemical shift values for different types of hydrogens in the molecules. They also display the effects of signal "splitting" occurring between hydrogen atoms attached to adjacent covalently-bonded carbon atoms. The splitting occurs because the spin states of the hydrogen nuclei a re coupled through the three bonds by which they are linked. This through-bond spin-spin coupling, or J-coupling, is responsible for the doublets, triplets, quartets, etc., of peaks for a given type of proton in the molecule, depending on the number of neighboring hydrogens.
Once we go beyond small organic molecules and acquire 1H-NMR spectra for polypeptides of the size of a small protein, we find that it is quite crowded and uinterpretable in detail. One can atrribute peaks in different regions to the characteristic types of hydrogen nuclei in polypeptides: amide hydrogens generally lie furthest downfield in the spectra (δ lies in the range 7 - 8.3), then aromatic hydrogens (δ 5.7 - 7.5), followed by hydrogens attached to Cα(δ 3.3 - 5.3), and finally other aliphatic hydrogens the furthest upfield (hydrogens attached to secondary Cβ, Cγ, or Cδ, δ 1.5 - 3.3; methyl group hydrogens, δ up to 1.5). Yet is is impossible to resolve all the peaks, let alone assign each specific hydrogen type in all the peptide's residues to a peak in the spectrum.
One approach to improve resolution for larger molecules is to employ a stronger magnetic field. Certainly the use of more powerful magnets has helped make NMR of polypeptides and small proteins useful. The implementation of multidimensional methods was also crucial. An analogy to the abilty of two-dimensional electrophoresis to resolve the hundreds or even thousands of individual proteins in a complex sample such as a cell extract is perhaps an apt one. A two-dimensional COSY (COrrelation SpectroscopY) spectrum probes the chemical shift of and J-coupling among the hydrogen nuclei, and makes possible the discernment of patterns of off-diagonal "cross-peaks" that are characteristic of the through-bond coupling within different types of amino acid side chains.
Another type of interaction becomes relevant in the application of NMR to macromolecules with well-defined conformational ensembles. This is the effect that nearby nuclei can have on each other's resonant frequency when they are simply juxtaposed "through space", and not linked by a small number of chemical bonds. This is the so-called nuclear Overhauser effect (nOe), which is effective only over very short ranges (the nuclei must be within about 5 Å). Multidimensional NMR is able to incorporate the effects of such through-space nuclear coupling, and such nOe signals can be similarly identified with cross-peaks in a 2-D spectrum.
Still, the "assignment problem" (i.e. answering the question of which peak belongs to which proton) in multidimensional NMR is the biggest challenge in applying it to the problem of protein structure determination. Very many methods have been developed to meet this challenge, which include using three- and even four-dimensional spectra and the labelling of the proteins being investigated (incorporation of isotopes) with 15N and 13C, so that the NMR spectra for these nuclei can be used as additional dimensions. The "editing" of polypeptide 1H-NMR spectra is also performed by incubation in solvent containing D2O, allowing for the exchange of labile hydrogens with deuterium (D).
In principle, solving the assignment problem with a sufficient quantity of nOe data allows the application of a set of distance constraints derived from the latter to calculate a family of closely related structures consistent with those constraints. Such a family of structures, called an "ensemble", is the form in which the results of structure determination by NMR are reported, although it is also common for a "most representative structure" to be derived from the ensemble.
Limitations of NMR
NMR and crystallography can be viewed as complimentary methoods in structure determination. Structures determined by these methods are most often in close agreement. While one cannot directly assign a resolution to an NMR protein structure, it does provide evidence of the dynamic features of proteins that is largely lacking in crystal structures. Some of the limitations of NMR as a protein structure determination method include the following:
- Upper limit on size (in VVP4e, given as 100 kD)
- NMR structures are not “high-resolution"
- “Dynamic” features may be hard to distinguish from uncertainty in data
- The protein must be soluble to high concentration