Lecture 9. Proteins: Secondary structure
Monday 26 September 2016
Characteristics of the peptide group. Defining polypeptide main-chain conformations: Dihedral angles (ω, φ, ψ) and Ramachandran plots. Secondary structure: α helix and β sheet. Secondary structure connections: turns, motifs. Fibrous protein structure: The α-helical coiled coil and keratin. Secondary structure propensities of amino acid residues.
Reading: VVP4e - Ch.6, pp.127-141.
Summary
Structural hierarchy: We discuss the nature of primary, secondary, tertiary and quaternary structure. Primary structure relates to the covalent description of a (possibly modified) polypeptide. Secondary structure is a "local" (in the sense of actual three-dimensional space) description of the conformation of a polypeptide chain. Secondary structures show regular patterns, the two prominent examples being the α helix (alpha helix) and the β sheet (beta sheet).
Tertiary structure is the overall shape of the polypeptide chain, which depends on the conformation of all the main chain and side chain bonds of the molecule. Quaternary structure refers to the cases in which separate polypeptide chains ("monomers" or "subunits") associate - usually noncovalently - to form "multimers" or "oligomers".
After an introductory review of hierarchy in protein structure, we take a more detailed look at the peptide bond and its chemical and structural characteristics. In order to understand and appreciate the conformations of polypeptide chains, we must also have a clear understanding of bond angles and how torsion angles (also called dihedral angles) are defined.
The peptide bond
Peptide bonds are the amide bonds that link amino acids together to form peptides (short chains of amino acids) and proteins (longer polypeptide chains that fold into specific three-dimensional conformations with biological function). The outstanding features of peptide bonds are described below:
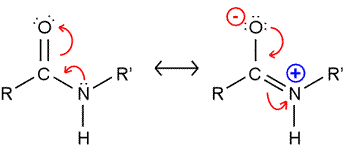
The peptide bond presents a significant barrier to rotation. The resonance structure on the right (see figure) is invoked to explain this, and bond length comparisons are consistent with partial double bond character. As a consequence, the atoms shown in the figure are all constrained to lie in the same plane. (Note that in polypeptide chains, R and R' represent Cα atoms and their attached groups.)
Peptide bonds: trans and cis forms
The peptide bond as we've seen is planar. But the planar conformation can be accommodated in two alternate forms denoted as trans and cis. The cis form is less stable because of its greater steric repulsion between the Cα atoms and their attached groups. Therefore, trans peptide bonds predominate in proteins.
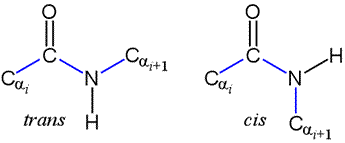
Note that a Newman projection of the C-N bond would show two dihedral angles: one in which the Cα atoms are anti (180°), corresponding to trans, and one in which the Cα atoms are eclipsed (0°), which is cis. This dihedral angle is called omega (ω).
X-Y and X-Pro peptide bonds, trans- and cis- forms.
Polypeptide chains
A polypeptide main chain, or "backbone" is made up of a repeating three-atom unit: the amide nitrogen (often denoted simply as N in this context), the alpha carbon (Cα), and acyl carbon (C′, or sometimes just C). The repeating functional groups, amide and acyl, give the main chain a polar character. The main chain amide (-NH- group) is a hydrogen bond donor, and the main chain acyl group (>C=O group) is a hydrogen bond acceptor.

The structural representation of a six-residue polypeptide segment shown above is meant to be a stereochemically correct representation of L-amino acids linked by trans peptide bonds (which are intersected by the dashed vertical lines).
The conformational space of a polypeptide main chain can be represented as an ordered set of points on a two-dimensional plot. The dihedral angles phi (φ) and psi (ψ) are both defined over a range of [−180°, 180°]. If the values for each residue are plotted, with φ along the horizontal axis, and ψ the vertical, this produces what is referred to as a Ramachandran plot.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Secondary structures: the α helix and β strand conformations
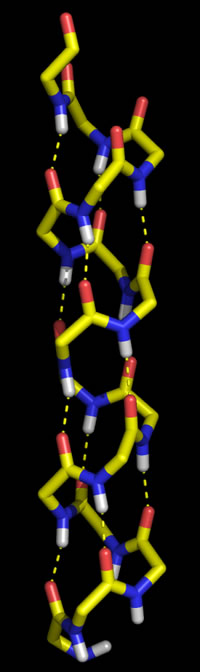
Polypeptide chains can adopt regularly repeating main chain conformations. Such conformations are generally the most stable (energetically favorable). One of these conformations, proposed first by Pauling and in fact observed prominently in protein tertiary structure, is the α helix (alpha helix, model at right).
Right: Molecular graphics image of 16 residues of poly Gly in the α helix conformation. In the stick representation, atoms are at ends and vertices, and yellow indicates carbon, red oxygen, blue nitrogen, and white hydrogen (Only main chain amide hydrogen atoms are shown). Hydrogen bonds are indicated by the dashed yellow lines, and form between the acyl oxygen of residue i (acceptor) and the amide group of residue i + 4 (donor).
The other regular conformation is the β strand conformation, Together with turns and loops, the α helix and β strand could be likened to "elements" that are put together to form higher-order "compounds" of of protein structure.
Characteristics of the α helix
The ideal α helix has the following features:
- Right-handed
- 3.6 residues per turn
- Pitch = 5.4 Å
- H-bond distances (amide N to acyl O) ~ 2.8 Å
- In proteins, an average length is ~ 12 residues (~ 18 Å)
β strands and sheets
Compared to the α helix, the β strand conformation yields a much more extended main chain. A fully-extended main chain would correspond to both φ and ψ dihedral angles close to ±180°. While this extension forecloses the possibility of intrachain interactions between main chain groups, it permits hydrogen bonding between these groups in juxtaposed β strands. The strands can align in either parallel or anti-parallel orientation, and juxtaposition of more strands gives rise to what are termed β sheets.
Connecting 2° structure elements: Turns
The architecture of globular proteins tends to be characterized by secondary structural elements that crisscross or traverse the molecule, and connecting segments where the direction of the polypeptide chain reverses, and the next 2° structure segment leads back toward the center of the molecule. These connecting segments - short turns and longer loops - thus tend to occur at the surface of the globular protein. Often such turn or loop segments are quite important for the protein's function.
Short turns, where the direction of the main chain changes radically, can be quite varied and show unusual conformations, although some forms recur quite frequently in protein structures. The most common short turns are well exemplified in β-hairpins (see below), in which two adjacent antiparallel β strands are linked.
 Left: Molecular graphics image of a type I beta turn. Residue positions i
through i + 3 are labeled, and H-bonds between main chain groups of i and i + 3 are
indicated by the dashed lines. The four-residue sequence for this turn is ESMG (residue i is Glu and
residue i + 3 is Gly). The view here is similar to that shown in Fig. 6-14 of VVP4e (p.136).
Left: Molecular graphics image of a type I beta turn. Residue positions i
through i + 3 are labeled, and H-bonds between main chain groups of i and i + 3 are
indicated by the dashed lines. The four-residue sequence for this turn is ESMG (residue i is Glu and
residue i + 3 is Gly). The view here is similar to that shown in Fig. 6-14 of VVP4e (p.136).
The type defined by four residues (designated i, i + 1, i + 2, and i + 3) and called β (beta) turns, or reverse turns, (also called β bends in our text), fit the following pattern: The acyl oxygen of residue i makes a hydrogen bond with the amino group of residue i + 3, while main chain groups of the central residues (i + 1 and i + 2) are not involved in the H-bonding pattern of an antiparallel β sheet. While four subtypes of β turns have been described, for most commonly observed of them glycine predominates in residue position i + 3, and Pro in position i + 1. Furthermore, Asp, Asn, and Ser are common at position i, since their side chains can make favorable interactions (H-bonds) with main chain groups of the central residues.
In the less-common γ (gamma) turn, the acyl oxygen of residue i makes a hydrogen bond with the amino group of residue i + 2.
Simple motifs and topological representations

The figure at right illustrates two simple ways in which β-strand elements can be connected. This more abstract way of representing protein structural features is called a topology diagram. Note that the β-hairpin motif aligns the connected β-strands in an antiparallel orientation, whereas an α-helix (or an extended loop) can be used to connect β-strands in a parallel fashion, by forming a β-α-β motif.
These very simple motifs can be repeated and elaborated into larger motifs, or supersecondary structure, as we'll see in the next lecture.
"Helical wheel" diagram and amphipathic helices
In terms of primary structure, β sheets can be made up of nonlocal β strand alignments. Beta strands from different polypeptide chains can be juxtaposed to form a sheet that is continuous across a subunit interface in a quaternary structure. Thus, we begin to visualize how secondary structure "elements" build up higher level structure through nonlocal interactions. Switching our focus to α helices, we can observe nonlocal associations between helices that build up higher level protein structure. In contrast to β sheets, in which β strand associations are made vis H-bonding between main chain polar groups, α helices associate due to favorable contacts between side chains.

The figure at left shows the positions of the side chains along a regular α-helix. For a right-handed helix with 3.6 residues per turn, rotation is clockwise as the polypeptide chain is followed N to C, and the 5th residue ends up in a position 40° clockwise relative to residue 1. Note that over a number of turns of the helix (21 residues or nearly six turns of the helix), a pattern of distribution of the side chains emerges in this example. Nonpolar side chains group on one side of the helix, whereas polar and charged side chains are grouped to the other side. This gives the helix an amphipathic character, having obvious implications for the orientation of such a helix in the context of tertiary structure. The nonpolar face of the helix can interact favorably with (and form part of) the nonpolar core of a globular protein, while the hydrophilic face will orient toward the surface.
"Helical wheel" diagram and amphipathic helices
As the example of an amphipathic regular α helix above suggests, the interaction of an α helix with other elements of protein structure occurs mainly through its side chain contacts. A specific example of this is the coiled coil motif, which occurs in many structural proteins such as keratin and an important class of transcription factors known as bZIP transcription factors. If an α helix is slightly distorted so that it has 3.5 residues per turn, then every seventh side chain will emanate from the same angular position on the helix surface (translated, of course, parallel to the helix axis, by 10 Å or so). This is known as a heptad repeat, and the energy required to distort the helix from the ideal geometry is made up for by favorable interactions between side chains in two or more such helices.
 Right: A helical wheel diagram for two heptad repeats that form a coiled-coil. The dashed lines indicate packing interactions involving the side chains at the a and d positions.
Right: A helical wheel diagram for two heptad repeats that form a coiled-coil. The dashed lines indicate packing interactions involving the side chains at the a and d positions.
A dimeric interaction (between two helices) is mediated by nonpolar side chains in two positions of the respective heptads, as shown in the figure. The heptad positions are labeled a - g, by convention, and it is the residues at positions a and d that form favorable contacts. The distortion of the two helices results in the formation of a superhelix - that is, the two helices wind around each other - hence the name "coiled-coil" for this type of motif.
Keratin
Two important examples of fibrous proteins, which tend to perform structural roles, are keratin and collagen. Let us first examine keratin, which is based on the coiled-coil motif just introduced above. The keratin polypeptide sequence features long stretches of heptad repeats. As a result, two keratin polypepide chains form α helices that wrap around each other, the axis of each forming a left-handed supercoil. The extended coiled-coils of keratin form higher order structures. This includes cross-linking of cysteine residues via disulfide bonds.
Collagen is a triple helix
Collagen is the principal protein in connective and other fibrous tissues, and is in fact the most abundant protein in humans. The primary structure of the collagen triple helix consists of a repetitive sequence, Gly-X-Y, where X and Y are often Pro or Hyp (hydroxyproline). This forms a left-handed helix with about three residues per turn, and we emphasize that this is not to be confused with the α helix! Three such left-handed helical chains wind around each other, the axis of each tracing out a right-handed supercoil. The amide group of glycine and acyl oxygen of proline form interchain hydrogen bonds through the middle of the triple helix.
There are many forms of collagen, and higher-order structures, in which triple-helices (denoted as "microfibrils") assemble.
Posttranslational modifications are an especially interesting aspect to collagen. Two important modifications:
- Hydroxyproline (Hyp) - stabilizes the triple helix conformation of collagen
- Allysine residues are modified lysine residues which lead to covalent cross-linking between triple helices
The hydroxylation of proline and lysine residues of collagen is carried out by hydroxylation enzymes that rely on ascorbate (vitamin C) for their function. Absence of vitamin C from the diet of humans leads to the collagen defects underlying the disease scurvy.
Residue propensities
Secondary structure propensities for amino acid residues. The following data is reproduced from Creighton (1993).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||